- Research
- Open access
- Published:
Effects of dexmedetomidine and esmolol on systemic hemodynamics and exogenous lactate clearance in early experimental septic shock
Critical Care volume 20, Article number: 234 (2016)
Abstract
Background
Persistent hyperlactatemia during septic shock is multifactorial. Hypoperfusion-related anaerobic production and adrenergic-driven aerobic generation together with impaired lactate clearance have been implicated. An excessive adrenergic response could contribute to persistent hyperlactatemia and adrenergic modulation might be beneficial. We assessed the effects of dexmedetomidine and esmolol on hemodynamics, lactate generation, and exogenous lactate clearance during endotoxin-induced septic shock.
Methods
Eighteen anesthetized and mechanically ventilated sheep were subjected to a multimodal hemodynamic/perfusion assessment including hepatic and portal vein catheterizations, total hepatic blood flow, and muscle microdialysis. After monitoring, all received a bolus and continuous infusion of endotoxin. After 1 h they were volume resuscitated, and then randomized to endotoxin-control, endotoxin-dexmedetomidine (sequential doses of 0.5 and 1.0 μg/k/h) or endotoxin-esmolol (titrated to decrease basal heart rate by 20 %) groups. Samples were taken at four time points, and exogenous lactate clearance using an intravenous administration of sodium L-lactate (1 mmol/kg) was performed at the end of the experiments.
Results
Dexmedetomidine and esmolol were hemodynamically well tolerated. The dexmedetomidine group exhibited lower epinephrine levels, but no difference in muscle lactate. Despite progressive hypotension in all groups, both dexmedetomidine and esmolol were associated with lower arterial and portal vein lactate levels. Exogenous lactate clearance was significantly higher in the dexmedetomidine and esmolol groups.
Conclusions
Dexmedetomidine and esmolol were associated with lower arterial and portal lactate levels, and less impairment of exogenous lactate clearance in a model of septic shock. The use of dexmedetomidine and esmolol appears to be associated with beneficial effects on gut lactate generation and lactate clearance and exhibits no negative impact on systemic hemodynamics.
Background
Persistent hyperlactatemia during septic shock resuscitation is associated with very high mortality and has been considered a hallmark of impending tissue hypoxia [1–8]. Therefore, some current guidelines recommend targeting resuscitation at normalizing lactate levels [4]. In clinical practice, this is accomplished by increasing cardiac output (CO) with fluid loading and/or inodilators [8, 9].
The paradigm of hypoxic-generated lactate has been recently challenged [1, 5]. Indeed, serum lactate levels during septic shock resuscitation represent a balance between aerobic or anaerobic generation, and clearance by different tissues. Consistent translational research has shown that muscle lactate generation can be triggered by epinephrine through β2-receptor stimulation, a process denominated aerobic glycolysis [10, 11]. On the other hand, an impaired lactate clearance might be present even without obvious signs of liver ischemia [12, 13]. In a recent experimental study, we found an early and severe impairment of exogenous lactate clearance to 10 % of sham values 1 hour after endotoxic [lipopolysaccharide (LPS)] shock induction [13]. This finding was not explained by liver hypoperfusion as tested with different techniques [13], although the pathogenic mechanisms were not explored. It is important to emphasize that in this study we addressed real clearance, applying a kinetic modeling after an intravenous (IV) bolus of sodium lactate that describes its elimination from the body over a short period of time [13]. The term “lactate clearance” has somehow been erroneously used in medical literature because a decrease in serum lactate levels could be caused either by a decreased production or increased clearance, and the inverse is also true [13, 14].
On the other hand, an overwhelming adrenergic response could contribute to persistent hyperlactatemia. Excessive α-mediated vasoconstriction might hasten hypoperfusion particularly in the hepatosplanchnic region, consequently increasing gut anaerobic lactate production, and impairing hepatic clearance by reducing portal or intrahepatic microcirculatory blood flow or eventually through metabolic effects [15–21]. High epinephrine levels can exacerbate aerobic glycolysis [10, 11]. The increasing awareness of the toxicity of sustained hyperadrenergia in critical illness has led to exploring adrenergic modulation or blockade as potential targets to attenuate adverse hemodynamic, microcirculatory, metabolic, and pro-inflammatory effects of sympathetic overstimulation [22–26]. Indeed, adrenergic modulation with an α2-agonist such as dexmedetomidine (DEX) [27–34], or β-blockers like esmolol (ESM) [35–40] have demonstrated favorable effects on diverse physiological and clinical outcome parameters in septic shock, and also on some potential determinants of persistent hyperlactatemia.
However, most of the studies with adrenergic modulation or β-blockade have been performed after the initial resuscitation period, and it is not known if these therapies are tolerated earlier in the evolution. In addition, since hyperlactatemia is a fundamental target for septic shock resuscitation, and due to the adrenergic influence over its determinants, it could be relevant to explore the impact of adrenergic modulation or β-blockade at this level. To address this subject, we performed a controlled experimental study in an endotoxic sheep model [13] aimed at (1) determining the effects of DEX and ESM as compared to LPS-control animals on lactate production and exogenous lactate clearance; and (2) evaluating the hemodynamic tolerance of DEX and ESM in the very early phase of septic shock. We hypothesized that both dexmedetomidine and esmolol decrease lactate production and attenuate impairment in exogenous lactate clearance in this model.
Methods
The experimental design was performed in agreement with the Guide for the Care and Use of Laboratory Animals, 8th edition (2011), and with the approval of the Comité de Etica y Bienestar Animal of the Pontificia Universidad Católica de Chile (CEBA 12-031). We used a well-standardized model of LPS shock in sheep that induces a characteristic hyperdynamic profile [13]. Details of the experimental setup have been reported elsewhere and will be summarized below [13].
Our model addressed three of the major determinants of persistent hyperlactatemia: muscle lactate generation as potentially representing adrenergic-driven aerobic glycolysis through microdialysis and serum epinephrine levels; gut lactate production as potentially representing anaerobic generation through portal oxygen venous saturation and lactate levels; and clearance using the exogenous lactate clearance technique proposed by Levraut et al [12].
Animal care and anesthesia
Sheep weighing 32 ± 5.2 kg were used. Animals were fasted 12 h before the experiments but with free access to water. Sheep were premedicated with 20 mg/kg ketamine and 0.25 mg/kg midazolam intramusculary. After inserting a peripheral intravenous line and injecting 30 mcg/kg fentanyl + 0.5 mg/kg atracurium + 1 mg/kg lidocaine intravenously, sheep were intubated and connected to mechanical ventilation in volume-control mode (Savina® 300, Dräger, Lübeck, Germany) with a tidal volume of 10 ml/kg. Anesthesia was sustained with a continuous infusion of midazolam, fentanyl, and ketamine. Muscle relaxation was maintained with a continuous atracurium infusion. During the surgical procedure, normal saline was infused at 10 ml/kg/h, and the rate was reduced to 5 ml/kg/h thereafter till the end of the experiment. Body temperature was kept at 38 ± 0.5 °C.
Surgery and instrumentation
An 8-Fr sheath was placed in both the left and right external jugular veins to advance pulmonary artery and hepatic vein catheters, respectively. The latter were positioned under ultrasound guidance. The left femoral artery and vein were surgically exposed, and arterial and central venous catheters were introduced for blood pressure monitoring, sampling, and administering fluids and drugs.
The abdomen was opened via a midline laparotomy and gastric contents were drained through a small gastrostomy. After this, the splenic vein was ligated and a portal catheter was placed for sampling. An ultrasound flow probe (Transonic, Ithaca NY, USA) was positioned around the hepatic artery and the portal vein to measure total hepatic blood flow, and the laparotomy was closed. A microdialysis catheter was inserted in the anterior quadriceps muscle (Harvard Apparatus, Holliston MA, USA).
Measurements
-
1.
Hemodynamic data were recorded every 30 minutes: heart rate (HR), mean arterial pressure (MAP), pulse pressure variation (PPV), and pulmonary arterial and occlusion pressures were measured with the standard procedure and displayed on a multi-modular monitor (GE Healthcare, Datex-Ohmeda, Madison WI, USA). CO was assessed by thermodilution (CO module, GE Healthcare, Datex-Ohmeda, Madison WI, USA).
-
2.
Systemic and hepatosplanchnic oxygen delivery and consumption: arterial, portal vein, hepatic vein, and mixed venous gases were assessed with a blood gas analyzer (i-Stat® bedside gas analyzer, Abbott Laboratories, Princeton NJ, USA). Total hepatic blood flow was measured with the ultrasound flow probe.
-
3.
Lactate assessment: serum lactate levels at every experimental time point were obtained from arterial and venous blood samples, and measured directly with a lactate scout monitor (Senslab, Leipzig, Germany). Measurements were performed in triplicate and results were averaged.
-
4.
Exogenous lactate clearance: lactate clearance was performed at the end of the experiment. An intravenous dose of sodium L-lactate (1 mmol/kg) was infused via the central venous catheter in 15 minutes [13]. Arterial blood samples were obtained at baseline and 1, 3, 6, 9, 15, and 20 minutes thereafter. Clearance was later calculated by the least squares technique with semi-logarithmic coordinates [13].
-
5.
Muscle microdialysis was performed using a CMA/402 microinjection pump at 0.3 μL/min of perfusion flow rate coupled to the microdialysis probe CMA/20 (Harvard Apparatus, Holliston, MA, USA). A 1-h equilibration period was allowed after insertion of the probe. The dialysate was collected into sealed 250 μL glass tubes in a refrigerated (4 °C) fraction collector (CMA142). Dialysate fractions for lactate assessment were collected during the last 30 minutes before time points A to D (Fig. 1, see below).
Fig. 1 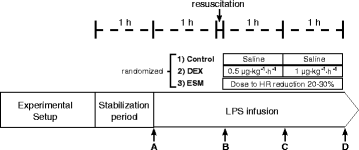
General scheme of the protocol. Complete hemodynamic, respiratory, and systemic and regional perfusion measurements were performed at points A, B, C, and D, except for lactate clearance that was performed at point D. LPS lipopolysaccharide, DEX dexmedetomidine, ESM esmolol, HR heart rate
-
6.
Epinephrine levels: serum epinephrine levels were determined in duplicate with an enzyme immunoassay kit (Rock Mountain Diagnostics, Colorado Springs, CO, USA).

Experimental protocol
After 1 h of postsurgical stabilization, basal measurements were performed (Fig. 1, point A). Then, septic shock was induced by administration of a 5 μg/kg LPS IV bolus (E coli 0111: β4; Millipore Sigma, St. Louis MO, USA) followed by a continuous LPS infusion at 4 μg/kg/h until the end of the experiment. During the first hour of LPS infusion no fluids or vasopressors were administered. Thereafter, resuscitation was performed with 5 ml/kg IV normal saline boluses repeated up to three times until a MAP of 55 to 60 mmHg or a pulse pressure variation below 10 % was achieved. If fluid loading failed to reestablish the MAP goal, norepinephrine (NE) was started at 0.1 mcg/kg/min and titrated in 0.1 mcg/kg/min increments every 5 minutes to the MAP target. Repeated series of measurements were performed after resuscitation (point B), and 1 and 2 h later (Fig. 1, points C and D, respectively). At each time point hemodynamic and respiratory variables, blood temperature, total hepatic blood flow, and arterial, mixed-venous, portal, and hepatic vein blood gases and lactate samples were taken simultaneously. Between points B and D, hypotension was basically managed with NE increments, but when the dose was increased by >0.5 mcg/kg/min during a 30-minute interval, and PPV was >15 % an additional 5 ml/kg fluid bolus was indicated.
After recording data set at point B, sheep were randomized into three groups of six animals each, LPS control, LPS-DEX (Precedex®, Hospira, Inc., Lake Forest, IL, USA), and LPS-ESM (Baxter Healthcare Corporation, Deerfield, IL, USA). To save animals and experiments we used only one LPS-control group against which DEX and ESM groups could be later compared. DEX was administered in two fixed doses for a 1-h period each (after point B, 0.5 μg/kg/h and after point C, 1.0 μ/kg/h). ESM was started at 15 mg/h and titrated every 5 minutes to achieve a reduction of 20–30 % in relation to HR at point B. This HR target was maintained until point D. Animals were euthanized with thiopental at the end of the experiment.
Statistics
All data are presented as mean ± standard deviation (SD). Statistical testing was two-sided and used the 5 % significance level.
We calculated that six animals would be required per arm to detect a significant difference of 1.8 mmol/l in serum lactate levels, with a sigma of 1.0 mmol/l, between LPS-control and LPS-DEX or LPS-ESM groups, assuming a type I error rate of 5 % and a power of 80 %. This calculation was based upon data from a previous study in which after 150 minutes of evolution, LPS animals presented a mean lactate of 3.7 + 0.9 mmol/l as compared with 1.3 + 0.3 mmol/l in controls [41].
The Shapiro-Wilk test was used to test for normality, with p values > 0.10 indicating a normal distribution. Comparisons of different time points within a single group (A, B, C and D) were performed by using Friedman’s test with Bonferroni’s post hoc correction. Comparisons of continuous variables between two groups were conducted with the t test or the Mann-Whitney U test, as appropriate.
Since the main objective of our study was to explore the individual impact of two anti-adrenergic interventions on the main determinants of persistent hyperlactatemia, we only performed comparisons between LPS-control and LPS-DEX groups, and between LPS-control and LPS-ESM groups, but not between the study drugs. Statistical analysis was performed using STATA version 11.0 (StataCorp LP, College Station, TX, USA).
Results
LPS induced a progressive septic shock with systemic hypotension, pulmonary hypertension and increasing NE requirements without differences between groups (Tables 1 and 2). DEX and ESM were well tolerated, and not associated with any adverse hemodynamic effect in terms of CO, mixed venous O2 saturation (SvO2), mixed venous-arterial carbon dioxide partial pressure gradients (p(v-a) CO2) or changes in NE requirements as compared to LPS controls (Tables 1 and 2). As expected LPS-ESM sheep exhibited a significant lower HR than LPS controls. The mean ESM dose was 26.7 ± 16.7 mg/h at point C and 32.2 ± 33.9 mg/h at point D.
LPS induced a progressive hyperlactatemia in the three groups, although serum lactate levels at point C were lower with DEX and ESM as compared to controls. This effect was maintained for ESM at point D (Table 3). DEX group exhibited significant lower epinephrine levels as compared to controls at point D (Table 3). No difference in muscle lactate production between groups was observed.
Portal lactate levels increased over time in all animals, but both DEX and ESM groups presented significant lower portal lactate values at points C and D as compared to LPS controls (Table 4). Exogenous lactate clearance was significantly higher in both DEX and ESM groups at point D (LPS-control 2.43 ± 1.14, LPS-DEX 6.97 ± 1.60, LPS-ESM 7.32 ± 2.2 ml/kg/min; p < 0.05) (Table 3 and Fig. 2). Total hepatic blood flow was comparable between groups (Table 4 and Fig. 2).

Comparison of dexmedetomidine, esmolol and controls in exogenous lactate clearance (A), cardiac output (B), and total hepatic blood flow (C) at the end of experiments. Both DEX and ESM were associated with less impairment in lactate clearance when compared to controls despite comparable systemic and regional hemodynamics. DEX dexmedetomidine, ESM esmolol
Discussion
The use of dexmedetomidine and esmolol was associated with lower arterial and portal lactate levels, and less impairment of exogenous lactate clearance in a model of septic shock. Both drugs were well tolerated when started very early after shock induction. DEX and ESM appear to be associated with beneficial effects on gut lactate generation and exogenous lactate clearance, and exhibit no negative impact on systemic hemodynamics.
Dexmedetomidine, an α2-agonist, attenuates sympathetic response to stress, and lowers epinephrine levels without adverse consequences on tissue perfusion [27–34]. Several experimental studies have consistently found anti-inflammatory effects and improvement in microcirculatory flow [32–34]. The drug is relatively well tolerated in anesthetized or critically ill patients, and even a post hoc analysis of the MENDS trial suggests an impact on mortality in septic patients [31]. Therefore, it could be useful as an adrenergic modulator in this setting.
On the other hand, the supporting evidence for β-blockers in sepsis is weak. Some small experimental and clinical studies have shown favorable effects on HR and hemodynamic or perfusion parameters, and also in inflammatory and metabolic parameters, particularly with nonselective blockers since most of these latter effects are β2 mediated [35]. However, β1 blockade could also exert anti-inflammatory effects, as was demonstrated by Hagiwara et al in a LPS rat model, on which an ultrashort-acting β-blocker inhibited nuclear factor-kappa B activity and attenuated histological lung damage [42]. Recently, a growing interest in esmolol, a short-acting selective β1-blocker has arisen mainly because of its pharmacokinetic characteristics [36–39]. An elegant experimental septic shock study found that ESM improves cardiac contractibility and vascular reactivity probably in relation to an anti-inflammatory effect [37]. In a randomized controlled study in stable septic shock patients, ESM reduced heart rate, decreased fluid requirements and lactate levels, and surprisingly showed a significant effect on mortality [38].
A disproportionate sympathetic response can be detrimental to critically ill patients as was demonstrated decades ago in another context such as chronic heart failure [22, 26]. Therefore, a growing interest in adrenergic modulation has arisen [22–26]. The big dilemma is to what extent can adrenergic modulation or blockade be accomplished without affecting basic survival responses especially in systemic hemodynamics. We found that both DEX and ESM appear to be well tolerated when started very early after shock onset, not only in terms of CO, MAP or NE requirements, but also from a metabolic point of view since both SvO2 and p(v-a)CO2 were comparable to LPS-controls. Furthermore, DEX and ESM were associated with favorable effects on both lactate generation and clearance as will be commented upon below. The few septic shock studies, in which ESM was assessed, started the drug hours after initial stabilization [38, 40]. In the case of DEX, this drug is not frequently used for primary sedation in septic shock patients due to the risk of inducing hemodynamic instability. DEX and clonidine might have opposite actions on vasomotor tone, a direct vasopressor, and indirect vasodilatory effects, with variable impact on MAP. When administered in healthy volunteers, DEX exerts a biphasic response, an initial increase in MAP due to stimulation of postsynaptic α2b receptors followed by a long-lasting fall in MAP due to its central sympatholytic action with a decrease in epinephrine and NE blood levels [43]. Some investigators have tested the hypothesis that central sympaticolysis might help to restore adrenergic vasoconstrictor responsiveness in septic shock by reversing downregulation of alpha receptors secondary to high endogenous catecholamines, and some experimental data tend to support this as feasible [27, 44]. However, this effect might take longer time. In any case, the hemodynamic tolerance exhibited by both drugs in our study opens new opportunities for research in this relevant subject.
Septic shock triggers a strong compensatory sympathetic activation with a wide array of circulatory, metabolic and immune effects that could potentially impact lactate production or clearance [22, 26]. Among metabolic effects, epinephrine stimulates aerobic glycolysis in skeletal muscle cells through β2 stimulation. This process generates and releases lactate into the systemic circulation as a metabolic fuel [10, 11]. A dysregulated sympathetic stress response or exogenous catecholamines could also impair hepatosplanchnic or microcirculatory flow at the gut or the liver through excessive vasoconstriction, triggering anaerobic lactate generation and potentially impairing hepatic lactate clearance [15–21]. We designed our study to address three potential sources for persistent hyperlactatemia on which an overactive sympathetic response could exert some influence. DEX induced a 37 % reduction in serum epinephrine levels, but noteworthy, this was not associated to any negative effect, neither on hemodynamics, nor in muscle lactate outflow. Muscle lactate production can be decreased experimentally by different approaches and inversely, exogenous β2-adrenergic stimulation with epinephrine and other β2-agonists increases aerobic lactate generation [10, 11]. In this latter case, the threshold over which epinephrine might hasten muscle lactate outflow is unknown but clearly DEX in relatively high doses did not affect this process.
The effects on the hepatosplanchnic region are of particular interest. We observed that LPS animals treated with ESM and DEX exhibited less increase in portal lactate levels as compared with LPS controls. Additionally, portal venous O2 saturation decreased over time only in controls, whereas total hepatic blood flow tended to decrease in all groups. Progressive gut hypoperfusion eventually ameliorated by adrenergic modulation or blockade could explain these findings. Unfortunately, the study design does not allow us to affirm this with certainty since we did not measure mesenteric or mucosal microcirculatory flow directly. Clinical and experimental studies have yielded conflicting results on splanchnic lactate balance in sepsis [45–54]. While some studies report anaerobic lactate generation by the gut as regional flow decreases, other have minimized the contribution of gut-generated lactate to systemic hyperlactatemia, since most of this lactate would be normally cleared by the liver [45–54]. Nonetheless, if hepatic lactate clearance is simultaneously impaired, the systemic impact of gut-generated lactate might be higher.
In a previous study using the same model, LPS induced an early and severe impairment in exogenous whole body net lactate clearance that was not related to total liver hypoperfusion or evident biochemical dysfunction [13]. Indeed, the very low porto-hepatic vein lactate differences suggested at least a liver metabolic inability to handle increased lactate loads. The decrease in lactate clearance reached a 10 % of sham values at the end of the experiments [13]. In the present study, exogenous lactate clearance fell to extremely low levels in LPS-controls similarly than in our previous study, but this decrease was significantly attenuated both in DEX and ESM groups. The combined effects on gut perfusion and lactate clearance might explain the impact of DEX and ESM on serum lactate levels.
How can DEX and ESM actions decrease gut lactate generation or influence exogenous lactate clearance? We did not design this study as a mechanistic one, and therefore we can only speculate about the mechanisms. α2-agonists such as DEX can attenuate the sympathetic response to surgery, decreasing circulating catecholamine levels in at least 10 to 20 %, but in our LPS model it was almost 40 % [27, 28]. Interestingly, DEX might exert opposite actions on vasomotor tone, a direct vasopressor and indirect vasodilatory effects, with variable impact on MAP [27, 28]. However, some experimental studies have shown that α2 agonists could have predominantly favorable effects over the gut microcirculation [33, 34], a particularly vulnerable territory [55]. Yeh et al found that DEX prevented gut microcirculatory abnormalities induced by sympathetic activation after surgical stress in rats [33]. Miranda et al found a significant attenuation of capillary perfusion deficits with DEX in a LPS model [34]. Thus, it is possible that the favorable effect of DEX on portal lactate levels might be consequence of an attenuated adrenergic vasoconstriction on mesenteric or gut microcirculatory flow. It is more difficult to explain the effects of ESM since no direct vascular effect can be postulated. However, some experimental studies have shown protective vascular or microcirculatory effects potentially related to immunomodulation, or increased release of endothelial nitric oxide among other actions, but this should be confirmed by further research [35–38]. It is also well known that LPS can induce acute portal hypertension resulting in gut mucosal hypoperfusion [56] and that nonselective β-blockers might reduce portal hypertension, but this effect might not be extrapolated to β1-selective blockers. The favorable impact of DEX and ESM on exogenous lactate clearance can be hardly explained by hemodynamic effects, since only a small difference in total hepatic blood flow compared to controls was observed at the end of the experiments. Potential liver microcirculatory or cellular effects of DEX and ESM should be explored in future studies.
We acknowledge several limitations of our study. First, we did not assess directly gut or liver microcirculation, thus this precludes us establishing any conclusion on the microvascular effects of both drugs. Second, we did not evaluate immunological aspects or biomarkers, eventually missing the exploration of the impact of adrenergic modulation at this level in our model. Third, small differences in portal and hepatic vein lactate levels between ESM and controls were observed at baseline and after shock induction. Biological variability in response to surgical stress or LPS could explain this finding, but the strong differences still observed at points C and D support our conclusions. Fourth, since our study was not aimed at comparing DEX with ESM, but rather both drugs against LPS controls, we cannot formulate any conclusion concerning eventual superiority of one over the other. Finally, our study can be considered only as hypothesis-generating and therefore these results should be confirmed and expanded in further research.
Conclusions
In conclusion, dexmedetomidine and esmolol were well tolerated and associated with lower arterial and portal lactate levels, and less impairment of exogenous lactate clearance in a model of endotoxic shock. Adrenergic modulation or blockade appears to be associated with beneficial effects on gut lactate generation and clearance, and exhibits no negative impact on systemic hemodynamics at least within the limits of our experimental model.
Abbreviations
CO, cardiac output; DEX, dexmedetomidine; ESM, esmolol; HR, heart rate; IV, intravenous; LPS, lipopolysaccharide; MAP, mean arterial pressure; NE, norepinephrine; PPV, pulse pressure variation; p(v-a)CO2, venous-arterial carbon dioxide partial pressure gradient; SvO2, mixed venous oxygen saturation
References
Hernandez G, Bruhn A, Castro R, Regueira T. The holistic view on perfusion monitoring in septic shock. Curr Opin Crit Care. 2012;18:280–6.
Hernandez G, Castro R, Romero C, de la Hoz C, Angulo D, Aranguiz I, et al. Persistent sepsis-induced hypotension without hyperlactatemia: is it really septic shock? J Crit Care. 2011;435:e9–14.
Cecconi M, De Backer D, Antonelli M, Beale R, Bakker J, Hofer C, et al. Consensus on circulatory shock and hemodynamic monitoring. Task force of the European Society of Intensive Care Medicine. Intensive Care Med. 2014;40:1795–815.
Dellinger RP, Levy MM, Rhodes A, Annane D, Gerlach H, Opal SM, et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013;39:165–228.
Garcia-Alvarez M, Marik P, Bellomo R. Sepsis-associated hyperlactatemia. Crit Care. 2014;18:503.
Hernandez G, Luengo C, Bruhn A, Kattan E, Friedman G, Ospina-Tascon GA, et al. When to stop septic shock resuscitation: clues from a dynamic perfusion monitoring. Ann Intensive Care. 2014;4:30.
Ospina-Tascón GA, Umaña M, Bermúdez W, Bautista-Rincón DF, Hernandez G, Bruhn A, et al. Combination of arterial lactate levels and venous-arterial CO2 to arterial-venous O2 content difference ratio as markers of resuscitation in patients with septic shock. Intensive Care Med. 2015;41:796–805.
Jansen TC, Van Bommel J, Schoonderbeek FJ, Sleeswijk Visser SJ, Van der Klooster JM, Lima AP, et al. Early lactate-guided therapy in intensive care unit patients: a multicenter, open-label, randomized controlled trial. Am J Respir Crit Care Med. 2010;182:752–61.
Hernandez G, Bruhn A, Luengo C, Regueira T, Kattan E, Fuentealba A, et al. Effects of dobutamine on systemic, regional and microcirculatory perfusion parameters in septic shock: a randomized, placebo-controlled, double-blind, crossover study. Intensive Care Med. 2013;39:1435–43.
Levy B, Perez P, Gibot S, Gerard A. Increased muscle-to-serum lactate gradient predicts progression towards septic shock in septic patients. Intensive Care Med. 2010;36:1703–9.
Levy B, Desebbe O, Montemont C, Gibot S. Increased aerobic glycolysis through beta2 stimulation is a common mechanism involved in lactate formation during shock states. Shock. 2008;30:417–21.
Levraut J, Ciebiera JP, Chave S, Rabary O, Jambou P, Carles M, et al. Mild hyperlactatemia in stable septic patients is due to impaired lactate clearance rather than overproduction. Am J Respir Crit Care Med. 1998;157:1021–6.
Tapia P, Soto D, Bruhn A, Alegría L, Jarufe N, Luengo C, et al. Impairment of exogenous lactate clearance in experimental hyperdynamic septic shock is not related to total liver hypoperfusion. Crit Care. 2015;19:188.
Vincent JL. Serial blood lactate levels reflect both lactate production and clearance. Crit Care Med. 2015;43:e209.
Spink WW, Reddin J, Zak SJ, Peterson M, Starzecki B, Seljeskog E. Correlation of plasma catecholamine levels with hemodynamic changes in canine endotoxin shock. J Clin Invest. 1966;45:78–85.
Moran JL, O’Fathartaigh MS, Peisach AR, Chapman MJ, Leppard P. Epinephrine as an inotropic agent in septic shock: a dose-profile analysis. Crit Care Med. 1993;21:70–7.
Minneci PC, Dens KJ, Banks SM, Costello R, Csako G, Eichacker PQ, et al. Differing effects of epinephrine, norepinephrine, and vasopressin on survival in a canine model of septic shock. Am J Physiol Heart Circ Physiol. 2004;287:H2545–54.
Levy B, Bollaert PE, Charpentier C, Nace L, Audibert G, Bauer P, et al. Comparison of norepinephrine and dobutamine to epinephrine for hemodynamics, lactate metabolism, and gastric tonometric variables in septic shock: a prospective, randomized study. Intensive Care Med. 1997;23:282–7.
De Backer D, Creteur J, Silva E, Vincent JL. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit Care Med. 2003;31:1659–67.
Meier-Hellmann A, Reinhart K, Bredle DL, Specht M, Spies CD, Hannemann L. Epinephrine impairs splanchnic perfusion in septic shock. Crit Care Med. 1997;25:399–404.
Di Giantomasso D, Bellomo R, May CN. The haemodynamic and metabolic effects of epinephrine in experimental hyperdynamic septic shock. Intensive Care Med. 2005;31:454–62.
Irving MH. Sympatho-adrenal hyperactivity--the key to irreversible shock? Postgrad Med J. 1969;45:523–9.
Dunser MW, Hasibeder WR. Sympathetic overstimulation during critical illness: adverse effects of adrenergic stress. J Intensive Care Med. 2009;24:293–316.
Hofer S, Steppan J, Wagner T, Funke B, Lichtenstern C, Martin E, et al. Central sympatholytics prolong survival in experimental sepsis. Crit Care. 2009;13:R11.
Rudiger A. Beta-block the septic heart. Crit Care Med. 2010;38:S608–12.
Andreis DT, Singer M. Catecholamines for inflammatory shock: a Jekyll-and-Hyde conundrum. Intensive Care Med. 2016. [Epub ahead of print].
Geloen A, Chapelier K, Cividjian A, Dantony E, Rabilloud M, May CN, et al. Clonidine and dexmedetomidine increase the pressor response to norepinephrine in experimental sepsis: a pilot study. Crit Care Med. 2013;41:e431–8.
Venn RM, Bryant A, Hall GM, Grounds RM. Effects of dexmedetomidine on adrenocortical function, and the cardiovascular, endocrine and inflammatory responses in post-operative patients needing sedation in the intensive care unit. Br J Anaesth. 2001;86:650–6.
Memiş D, Hekimoğlu S, Vatan I, Yandim T, Yüksel M, Süt N. Effects of midazolam and dexmedetomidine on inflammatory responses and gastric intramucosal pH to sepsis, in critically ill patients. Br J Anaesth. 2007;98:550–2.
Memiş D, Kargi M, Sut N. Effects of propofol and dexmedetomidine on indocyanine green elimination assessed with LIMON to patients with early septic shock: a pilot study. J Crit Care. 2009;24:603–8.
Pandharipande PP, Sanders RD, Girard TD, McGrane S, Thompson JL, Shintani AK, et al. Effect of dexmedetomidine versus lorazepam on outcome in patients with sepsis: an a priori-designed analysis of the MENDS randomized controlled trial. Crit Care. 2010;14:R38.
Taniguchi T, Kurita A, Kobayashi K, Yamamoto K, Inaba H. Dose- and time-related effects of dexmedetomidine on mortality and inflammatory responses to endotoxin-induced shock in rats. J Anesth. 2008;22:221–8.
Yeh YC, Sun WZ, Ko WJ, Chan WS, Fan SZ, Tsai JC, et al. Dexmedetomidine prevents alterations of intestinal microcirculation that are induced by surgical stress and pain in a novel rat model. Anesth Analg. 2012;115:46–53.
Miranda ML, Balarini MM, Bouskela E. Dexmedetomidine attenuates the microcirculatory derangements evoked by experimental sepsis. Anesthesiology. 2015;122:619–30.
Hamzaoui O, Teboul JL. The role of beta-blockers in septic patients. Minerva Anestesiol. 2015;81:312–9.
Jacquet-Lagrèze M, Allaouchiche B, Restagno D, Paquet C, Ayoub JY, Etienne J, et al. Gut and sublingual microvascular effect of esmolol during septic shock in a porcine model. Crit Care. 2015;19:241.
Kimmoun A, Louis H, Al Kattani N, Delemazure J, Dessales N, Wei C, et al. β1-adrenergic inhibition improves cardiac and vascular function in experimental septic shock. Crit Care Med. 2015;43:e332–40.
Morelli A, Donati A, Ertmer C, Rehberg S, Kampmeier T, Orecchioni A, et al. Microvascular effects of heart rate control with esmolol in patients with septic shock: a pilot study. Crit Care Med. 2013;41:2162–8.
Aboab J, Sebille V, Jourdain M, Mangalaboyi J, Gharbi M, Mansart A, et al. Effects of esmolol on systemic and pulmonary hemodynamics and on oxygenation in pigs with hypodynamic endotoxin shock. Intensive Care Med. 2011;37:1344–51.
Morelli A, Ertmer C, Westphal M, Rehberg S, Kampmeier T, Ligges S, et al. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. JAMA. 2013;310:1683–91.
Dubin A, Edul VS, Pozo MO, Murias G, Canullán CM, Martins EF, et al. Persistent villi hypoperfusion explains intramucosal acidosis in sheep endotoxemia. Crit Care Med. 2008;36:535–42.
Hagiwara S, Iwasaka H, Maeda H, Noguchi T. Landiolol, an ultrashort-acting beta1-adrenoceptor antagonist, has protective effects in an LPS-induced systemic inflammation model. Shock. 2009;31:515–20.
Dünser MW, Gradwohl-Matis I. α(2)-agonists to restore adrenergic vasoconstrictor responsiveness in septic shock: thinking outside of the box or fishing in the wrong pond? Crit Care Med. 2013;41:2838–40.
Lankadeva YR, Booth LC, Kosaka J, Evans RG, Quintin L, Bellomo R, et al. Clonidine restores pressor responsiveness to phenylephrine and angiotensin II in ovine sepsis. Crit Care Med. 2015;43:e221–9.
Mizock BA. The hepatosplanchnic area and hyperlactatemia: a tale of two lactates. Crit Care Med. 2001;29:447–9.
Hernandez G, Regueira T, Bruhn A, Castro R, Rovegno M, Fuentealba A, et al. Relationship of systemic, hepatosplanchnic, and microcirculatory perfusion parameters with 6-hour lactate clearance in hyperdynamic septic shock patients: an acute, clinical-physiological, pilot study. Ann Intensive Care. 2012;2:44.
Bellomo R, Kellum JA, Pinsky MR. Transvisceral lactate fluxes during early endotoxemia. Chest. 1996;110:198–204.
Michaeli B, Martinez A, Revelly JP, Cayeux MC, Chioléro RL, Tappy L, et al. Effects of endotoxin on lactate metabolism in humans. Crit Care. 2012;16:R139.
Severin PN, Uhing MR, Beno DW, Kimura RE. Endotoxin-induced hyperlacta- temia results from decreased lactate clearance in hemodynamically stable rats. Crit Care Med. 2002;30:2509–14.
Santak B, Radermacher P, Adler J, Iber T, Rieger KM, Wachter U, et al. Effect of increased cardiac output on liver blood flow, oxygen exchange and metabolic rate during long-term endotoxin-induced shock in pigs. Br J Pharmacol. 1998;124:1689–97.
Samsel RW, Cherqui D, Pietrabissa A, Sanders WM, Roncella M, Emond JC, et al. Hepatic oxygen and lactate extraction during stagnant hypoxia. J Appl Physiol. 1991;70:186–93.
Tashkin DP, Goldstein PJ, Simmons DH. Hepatic lactate uptake during decreased liver perfusion and hyposemia. Am J Physiol. 1972;223:968–74.
Naylor JM, Kronfeld DS, Freeman DE, Richardson D. Hepatic and extrahepatic lactate metabolism in sheep: effects of lactate loading and pH. Am J Physiol. 1984;247:E747–55.
Creteur J, De Backer D, Sun Q, Vincent JL. The hepatosplanchnic contribution to hyperlactatemia in endotoxic shock: effects of tissue ischemia. Shock. 2004;21:438–43.
Edul VS, Ince C, Navarro N, Previgliano L, Risso-Vazquez A, Rubatto PN, et al. Dissociation between sublingual and gut microcirculation in the response to a fluid challenge in postoperative patients with abdominal sepsis. Ann Intensive Care. 2014;4:39.
Brienza N, Ayuse T, Revelly JP, O’Donnell CP, Robotham JL. Effects of endotoxin on isolated porcine liver: pressure-flow analysis. J Appl Physiol (1985). 1995;78:784–92.
Acknowledgements
We acknowledge Carolina Henriquez RN for her technical support in the experiments
Funding
This study was supported by a FONDECYT Chile Grant project number 1130200.
Availability of supporting data
All the data are available for the editors and reviewers of Critical Care, and will be submitted if required.
Authors’ contributions
GH, AB, GO, and JB were responsible for the study concept and design, the analysis and interpretation of data, and drafting of the manuscript. PT, LA, CL, and JG performed the acquisition of data and contributed to the drafting of the manuscript. DS and LA conducted the blood determinations and contributed to the drafting of the manuscript. NJ, PA, RR, and PT performed the surgical preparation and contributed to the discussion. RC and EK developed and maintained the database, performed statistical analyses, helped in the drafting of the manuscript, and critically revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Authors’ information
No additional information is provided.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
All the coauthors approve the publication of this manuscript.
Ethical approval and consent to participate
The study was approved by the Comité de Etica y Bienestar Animal of the Pontificia Universidad Católica de Chile (CEBA 12-031).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hernández, G., Tapia, P., Alegría, L. et al. Effects of dexmedetomidine and esmolol on systemic hemodynamics and exogenous lactate clearance in early experimental septic shock. Crit Care 20, 234 (2016). https://doi.org/10.1186/s13054-016-1419-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13054-016-1419-x